Computational Studies on DNA Polymerase β
DNA polymerase β (pol β) fills short DNA gaps during repair synthesis in the base excision repair (BER) pathway and plays a role in chromosomal replication (Beard and Wilson, 1998). DNA pol β is composed of two domains: lyase (8-kDa) and polymerase (31-kDa). The shape of the latter is like a hand with fingers, palm, and thumb subdomains (Ollis et al., 1985). Structural and kinetic evidence suggests that pol β selects the correct deoxynucleoside triphosphate (dNTP) from a pool of structurally similar molecules by moving between open and closed states: substrate binding and product release occur in the open state, while the chemical DNA extension occurs in the closed state (Beard and Wilson, 1998).
Our group has collaborated with leading experts in the chemistry and biology of DNA polymerases to elucidate how these enzymes replicate and repair normal and damaged DNA. In particular, our group has established novel protocols to investigate fidelity mechanisms of mammalian pol β. These studies have revealed the conformational and chemical events that enable it to discriminate between correct and incorrect base pairs and hence control fidelity.
Slow local rearrangements in pol β's conformational pathway. Kinetic data reveal slow conformational steps before and after chemistry (steps 2 and 4 of the catalytic cycle in Fig. 1). Enzyme kinetic measurements, however, suggest that the subdomain opening/closing motions themselves are relatively fast (Vande Berg et al., 2001). Utilizing advances in modeling and methodology, we simulated the partial opening of pol β after chemistry (Yang et al., 2002) started from an intermediate structure. We proposed a sequence of events during pol β’s opening after chemistry: (1) Phe272 ring flips away from Asp192, (2) large thumb movement, and (3) Arg258 rotates toward Asp192. The rate constant we derived for the Arg258 rotation (~2 × 10-2 s-1) suggests that the Arg258 rotation is slow relative to subdomain motions (i.e., thumb opening/closing). Subsequent MD (Arora and Schlick, 2005) and transition path sampling (Radhakrishnan and Schlick, 2004b) simulations for the subdomain closing motions also corroborate that a large free energy barrier is associated with Arg258’s rotation. This work supports the hypothesis that, although chemistry may be the rate-limiting step in the overall insertion pathway, pol β’s conformational change directs assembly of the active site.
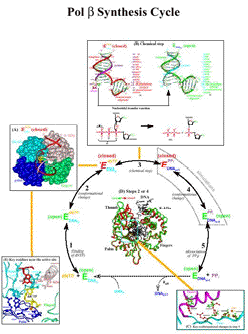
Figure 1. Schematic drawing of pol β's synthesis cycle.
Standard MD and transition path sampling studies of pol β’s conformational change provide evidence for induced-fit mechanism. In an induced-fit mechanism, the correct incoming nucleotide triggers the requisite conformational change while an incorrect incoming nucleotide hampers the process. Our simulations of pol β with and without the correct incoming nucleotide support this mechanism (Arora and Schlick, 2004; Arora and Schlick, 2005) (Fig. 2). Further support is provided by studies of pol β bound to several different mismatched base pairs (G:G, G:T, and T:T) using standard MD simulations (Arora et al., 2005) and transition path sampling (G:C and G:A) (Radhakrishnan and Schlick, 2005). Significantly, we were able to identify the multiple transition regions by a novel divide and conquer approach (Radhakrishnan and Schlick, 2004b) and the development of an efficient new free energy method (Radhakrishnan and Schlick, 2004a).
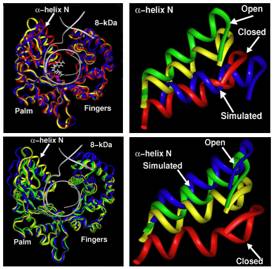
Figure 2: Ca traces of superimposed pol β/DNA complex with dCTP (top left) and without dCTP (bottom left) for the intermediate starting structure (yellow), crystal closed (red), and crystal open (green) and the trajectory final structures (blue) [Arora & Schlick, 2004].
Notable are the residue motions in the thumb subdomain and the 8-kDa domain. The positions of a-helix N in the simulated systems are compared to the crystal structures in panels on the right (top, with dCTP, and bottom, without dCTP)
The complex reaction profile delineated in (Radhakrishnan and Schlick, 2004b) reveals five transition-state regions –– partial thumb closing, Asp192 flip, Arg258 rotation, Phe272 flip, and rearrangement of catalytic region –– and relative free-energies involved (Fig. 3). Studies with the G:A (Fig. 4) mismatch suggest that the closed state (in contrast to the G:C case) is unstable, and that multiple pathways are involved in the overall pathway.
Recent work also revealed the important role of divalent ions in this process. Depending on ion positions, optimal and variant pathways can be identified (Li et al., 2014). Altered pathways of pol β in the presence of an oxidized nucleotide can also be identified experimentally snd computationally (Freudenthal et al., 2015, Kim et al., 2016).
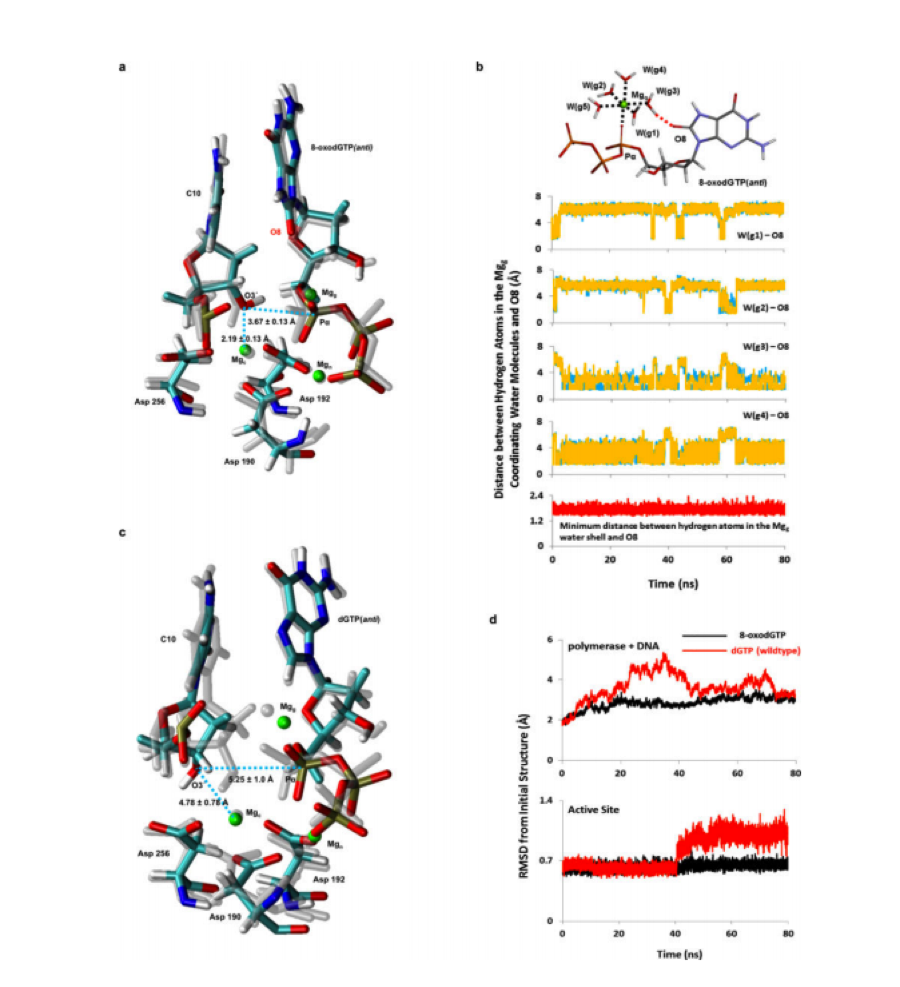
Figure 3: MD simulation analysis of 8-oxo-dGTP(anti) and dGTP(anti) opposite Cy. The catalytic (Mgc ), nucleotide (Mgn), and ground (Mgg) magnesium metal ions are shown in green, and average distances over the course of the simulation are indicated for Pα-O3’ and Mgc -O3’. Four of the five water molecules in the water shell (W(g1-g4)) contribute to hydrogen-bonding interactions with O8 (Freudenthal et al., 2015).
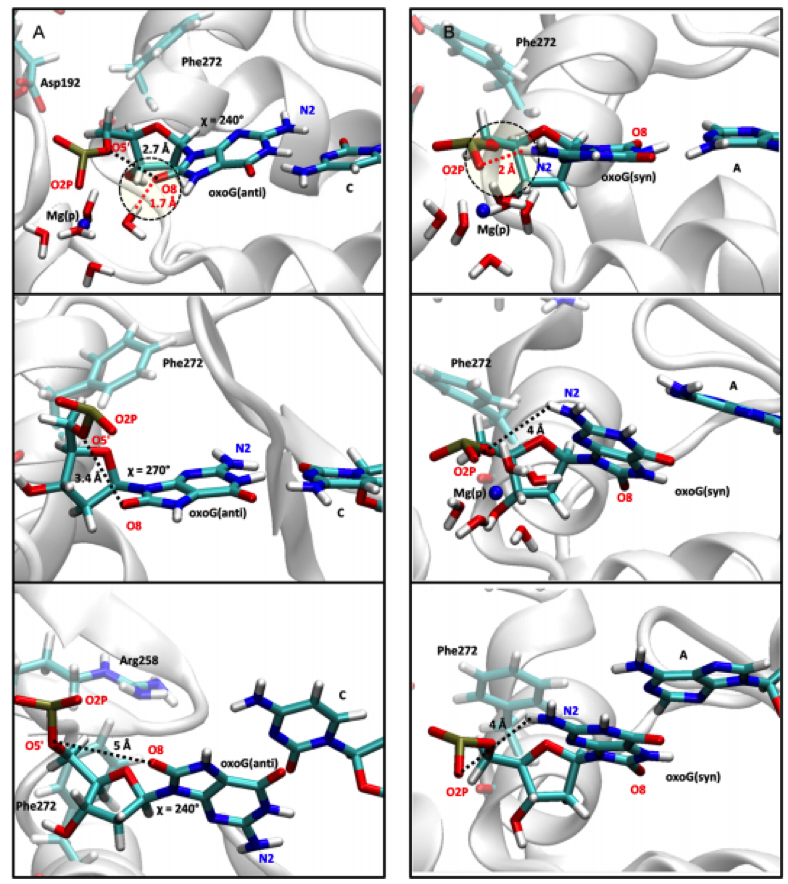
Figure 4: In the oxoG(anti):C system, the Mg(p) ion stabilizes the base pair interaction by reducing electrostatic repulsion between O8 of the guanine residue and the phosphate group and strengthening the hydrogen bonding interaction between a water molecule and O8 (top image). After the Mg(p) ion dissociates, both the distance between O8 and the phosphate group and the glycosyl angle χ increase so that the base pair interaction is destabilized (middle image). The base pair then breaks and the χ angle adjusts slightly (bottom image) (Kim et al., 2016).
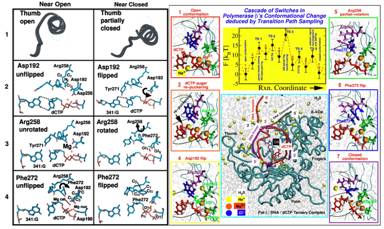
Figure 5. Left: Molecular snapshots near open (left column) and closed (right column) states of pol β for four transition state regions (Radhakrishnan and Schlick, 2004b): (1) Partial thumb closing. (2) Asp192 flip. (3) Arg258 partial rotation. (4) Phe272 flip. Right: Overall captured reaction kinetics profile (from TPS) for the conformational transition of pol β (for G:C) from open (state 1) to closed (state 7) forms showing free energies (in kBT) associated with the different transition state regions (Radhakrishnan and Schlick, 2004b). The metastable basins (in red) along the reaction coordinate are numbered 1-7. See animated sequence of this closing on our website: http://www.biomath.nyu.edu/index/gallery.html.
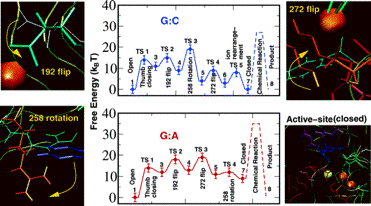
Figure 6. Overall captured reaction kinetics profile for pol β's closing transition followed by chemical incorporation of dNTP for G:C and G:A systems (Radhakrishnan and Schlick, 2005).
The barriers to chemistry (dashed peaks) are derived from experimentally measured kpol values. The profiles were constructed by employing reaction coordinate characterizing order parameters in conjunction with transition path sampling. The potential of mean force along each reaction coordinate is computed for each conformational event.
Pol β’s chemical mechanism for G:C vs. G:A systems. In addition to exploring the conformational pathways, we have studied pol β's mechanism for catalyzing the nucleotidyl-transfer reaction of correct and incorrect insertions using both quantum mechanics (QM) and combined quantum mechanics and molecular mechanics (QM/MM) approaches (Radhakrishnan and Schlick, 2006; Alberts et al., 2007; Bojin and Schlick, 2007). The proposed pathways for the G:C and G:A systems support a series of transient intermediates involving a Grotthuss hopping mechanism of proton transfer between water molecules and the three conserved aspartate residues in the enzyme’s active site (Radhakrishnan and Schlick, 2006) (Fig. 5).
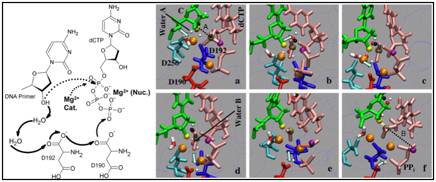
Figure 7. Left: Schematic drawing of the mechanism of concerted proton-hops during the pol β chemical reaction (Radhakrishnan and Schlick, 2006).
Right: Captured reaction intermediates associated with G:C incorporation (Radhakrishnan and Schlick, 2006). The colors represent: cyan (D256), red (D190), blue (D192), pink (dCTP), green (CYT: terminal DNA primer), black (the O3'H-proton), yellow (the O3' oxygen, attacking nucleophile), tan (central phosphorous), purple (leaving O3A oxygen), and orange (magnesium ions). The oxygens and hydrogens of water molecules are in red and white, respectively. The arrows denote the location and direction of proton hop.
The Revealed Conformational Pathway Explains Mutant Behavior
Remarkably, the behavior of the highly inactive E295K pol β mutant can be explained by this revealed pathway before chemistry. Unfavorable electrostatic and steric interactions in DNA pol β E295K mutant produce an altered pathway that correlates with the enzyme’s observed behavior (Li et al., 2012).
Evidence of Induced-fit Mechanism in African Swine Fever Virus Pol X
African Swine Fever Virus DNA pol X is another member of the X-family that appears to repair oxidative damage. However, pol X lacks the fingers subdomain. We investigated pol X’s interactions with its substrates by modeling pol X/DNA and pol X/DNA/dNTP complexes (Sampoli Benitez et al., 2006). This revealed that pol X/DNA complexes favor an open conformation distinct from the open conformation assumed by free pol X and pol X/DNA/dNTP complexes favor a more closed conformation. Thus, despite its structural differences, pol X seems to follow an induced-fit mechanism similar to pol β.
Our series of MD simulations of pol X bound to different mismatched base pairs [i.e., C:C, A:G, G:G (anti), and G:G (syn)] helped explain its differential binding affinity (Sampoli, Benitez et al., 2008). Intriguingly, our simulations provide an explanation why G:G is preferentially bound: in the G:G (syn) mismatch simulation, the thumb exhibits a large-scale conformational change from an open to closed state that is similar to what occurs with the correct G:C base pair; in contrast, the A:G and G:G (anti) systems only display partial thumb closing and the C:C system maintains the thumb open. Thus, only the G:G (syn) base pair fits well within the helix via a Hoogsteen base pair arrangement that is geometrically similar to standard Watson-Crick base pairs (see all mismatches in Fig. 6). Besides exploring pol X’s G:G preference, our work provided a geometric selection view that agrees with kinetics data for other mispairs. Similar studies also explain how pol X preferentially accommodates dATP opposite the common oxidative lesion 8-oxoguanine (Sampoli-Benitez et al., 2013).
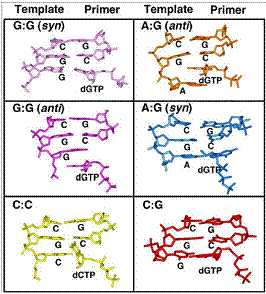
Figure 8. Geometry of template-primer DNA base pairs bound to pol X.
Complex Connections between Structure, Flexibility, and Fidelity in Pol λ
Pol λ is a low to moderate fidelity member of the X-family of DNA polymerases. Although pol λ and pol β share many similarities including a striking structural homology, the two enzymes have several divergent properties. Experimental evidence suggests that pol λ participates in the repair of double strand breaks through the non-homologous end-joining pathway and also may play a back up role for pol β in BER (Moon et al., 2007).
Pol λ’s conformational pathway. In agreement with X-ray crystal data (Garcia-Diaz et al., 2005), our MD simulations suggest that pol λ does not demonstrate large-scale subdomain movements like pol β (Foley et al., 2006). Significant DNA motion exists, however, as well as sequential side-chain motions associated with Arg514, Arg517, Ile492, Phe506, and Tyr505, all coupled to active-site divalent ions and DNA motion (Fig. 7). These motions transform pol λ to the chemistry-competent state. As proposed for pol β, these residue motions may serve as gate-keepers by controlling the evolution of the reaction pathway before the chemical reaction.
Pol λ’s slippage tendency. Pol λ’s propensity for single-base deletions is greater than pol β’s and even exceeds the error rates of Y-family enzymes (Bebenek et al., 2003; Garcia-Diaz and Kunkel, 2006). X-ray crystal data on pol λ suggests that these deletions occur through DNA template strand slippage (Garcia-Diaz et al., 2006). Our simulations of mutant derivatives of pol λ at position 517 indicate that discrete orientations of the mutant residues can occur and that these orientations impact protein-coupled DNA stability by forming unfavorable electrostatic interactions (Bebenek et al., 2008; Foley and Schlick, 2008). The extent of DNA movement in our simulations is mutant dependent and mirrors the deletion error rates already determined for wild-type pol λ and the Lys and Ala mutants (Bebenek et al., 2008). Our studies demonstrate that pol λ’s unique architecture may facilitate deletion errors because of the tenuous nature of these specific protein/DNA interactions compared to other DNA polymerases (Foley and Schlick, 2009; Foley et al., 2010).
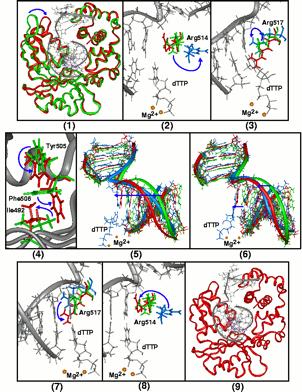
Figure 9. Proposed sequence of events for the catalytic cycle of DNA pol λ (Foley et al., 2006).
In all panels, the X-ray crystal binary (PDB entry 1XSL) and ternary (PDB entry 1XSN) complexes are green and red, respectively, and intermediate conformations of residues and DNA are blue. Blue arrows represent motion. The correct incoming nucleotide, here dTTP, is shown in all panels with both the catalytic and nucleotide-binding ions (gold spheres) except panel 4. Panel 1 depicts the closing motion of the loop containing ß-strand 8 in the thumb subdomain in response to binding of the correct incoming nucleotide, dTTP (purple); superimposed Catraces of X-ray crystal binary and ternary are shown. Panel 2 shows the start of Arg514's transition from the binary to the ternary position with the flip of Arg514 from the binary state position away from the DNA as indicated by the arrow. Panel 3 shows the partial flip of Arg517 from the binary to the ternary position. Panel 4 shows the flips of Ile492, Tyr505, and Phe506 from the binary to the ternary state positions. Panel 5 shows the DNA in transition from the binary to the ternary state which occurs after the flips of Ile492, Tyr505, and Phe506. Panel 6 shows the completed transition of the DNA to its ternary position. Panel 7 shows the completed flip of Arg517 from its intermediate position to the ternary position. Panel 8 shows the completed flip of Arg514 from its intermediate position to its ternary position. Panel 9 shows the closed active conformation of the pol λ/DNA complex with substrate (purple) and ions (gold spheres) bound.
Simulations of the Conformational and Chemical Pathways in Dpo4
Despite their low-fidelity, Y-family DNA polymerases are able to perform the very important task of bypassing lesions; that is, they can be loaded onto the replication fork to transverse bulky lesions and rescue replication when a replicative DNA polymerase is stalled. Sulfolobus solfataricus DNA Polymerase IV (Dpo4), a thermophilic archeal protein capable of translesion synthesis, is one of the few Y-family DNA polymerases crystallized and studied experimentally (Ling et al., 2001); see the structure of Dpo4 in Figure 8.
Figure 10. 3D structure of Dpo4.
The nature of the conformational changes in Dpo4. The high error rate of Y-family polymerases in replicating undamaged DNA and their ability to bypass DNA lesions may be partially due to a more open active site (relative to high-fidelity polymerases) that can even accommodate two template bases simultaneously (Ling et al., 2003). Through long-term MD simulations of the ternary Dpo4/DNA/dCTP closed system (with 8-oxoG bound in anti orientation to incoming C), we have captured significant rigid-body motions (Fig. 9) in the finger and little finger domains that facilitate DNA translocation (Wang et al., 2006). Compared to the thumb and little finger domains, the catalytic core of Dpo4, particularly the palm domain, is relatively rigid. The motions of the finger and little finger domains appear to push the nascent base pair as well as the DNA duplex away from the active site to make room for the next incoming nucleotide (see Fig. 9). We also found that the active site of Dpo4 does not undergo large changes upon binding of a correct incoming nucleotide as found in higher fidelity pol β.
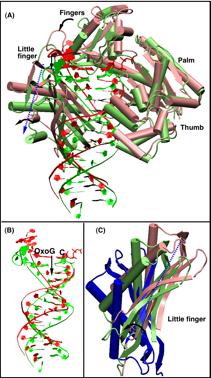
Figure 11. Captured structural changes of Dpo4 after chemistry complex following removal of metal ions and pyrophosphate (Wang et al., 2006).
(A) Protein and DNA changes before (red) and after (green) simulation; superimposition performed according to the palm subdomain. (B) Enlarged view of the DNA shift. (C) Comparison of the LF domains before (light red) and after (light green) simulation to that of the Dbh apo-structure (Silvian et al., 2001) (blue) by superimposing the palm domains.
The nucleotidyl transfer reaction in Dpo4. Our QM/MM studies of possible Dpo4 chemical reaction pathways for the correct insertion of dCTP opposite 8-oxoG revealed that the most favorable reaction path involves initial deprotonation of O3'H via two bridging water molecules to dCTP, overcoming an overall energy barrier of approximately 20.0 kcal/mol (Wang and Schlick, 2007; Wang and Schlick 2008). The proton then migrates to the γ-phosphate oxygen of dCTP as the nucleotide is joined to the primer terminus and pyrophosphate is formed. In contrast, initiating the chemical reaction from the less ideal state of the crystal structure requires a much higher activation energy barrier (29.0 kcal/mol) due to longer distances for O3'H proton transfer and distorted conformations of the proton acceptors. This significant difference demonstrates that the pre-chemistry reorganization in Dpo4 costs approximately 4.0 to 9.0 kcal/mol depending on the active-site environment. Compared to the higher fidelity pol β, Dpo4 has a higher chemical reaction barrier, which may result from its more solvent-exposed active site.
Conformational Transitions in Pol μ
Pol μ, like pol λ, is a pol x family member that also associates with the non-homologous end joining (NHEJ) pathway, where double stranded breaks are repaired. Like pol λ, pol μ can handle a gapped template, but unlike this family relative it can also handle a primer terminus unpaired with its template. Its unique associated pathway helps explain this enzyme’s remarkable properties (Li & Schlick 2010; Li & Schlick 2013).
Emerging Themes and Insights
Our studies of the conformational and chemical pathways in various DNA polymerases reveal how structure is intimately linked to function and the tradeoffs involved between high and low fidelity enzymes. High fidelity polymerases have evolved characteristics to facilitate specific cellular functions while the low fidelity characteristics of other polymerases allow them to tackle difficult tasks like bulk lesion bypass. Higher fidelity polymerases like pol β exhibit large-scale subdomain motions before and after chemistry. They also have geometrically restrictive active sites and little tolerance for DNA distortion. Lower fidelity polymerases have fast subdomain motions (e.g., pol X), no large-scale subdomain motions (e.g., pol λ), and/or extensive DNA motion in their catalytic cycle (e.g., pol λ and Dpo4). These enzymes are also more tolerant of active site or DNA distortion.
The emerging unifying theme is a relationship between cooperative conformational changes and chemical activation that define kinetic checkpoints in nucleotide incorporation and discrimination. We identified three distinct avenues (Fig. 10): conformational change upon substrate binding, a pre-chemistry evolution of the system to the reaction-competent state, and the transfer of nucleotides through chemistry. Utilizing the pre-chemistry avenue concept we can interpret different polymerase mechanisms by their differing pre-chemistry energy barriers. Data from structural and computational studies of several polymerases (e.g., pol β, pol λ, pol μ, pol X, and Dpo4) in binary and ternary states emphasize that polymerase conformational changes can vary significantly among polymerases, even within the same family. Thus, while polymerases may appear to lack a unified description of their wide variation in DNA synthesis and repair fidelities based solely on subdomain motions, the pre-chemistry concept offers a plausible mechanism to reconcile a number of observed differences in the fidelities of various polymerases.
Moreover, intrinsic motions of DNA polymerases underlie their remarkable specificity and selectivity through a hybrid induced fit/conformational selection mechanism (Foley et al., 2012). Variability in ion positions and inclusion of additional ions also create variability in overall pathways (Freudenthal et al., 2015; Kim et al., 2016).
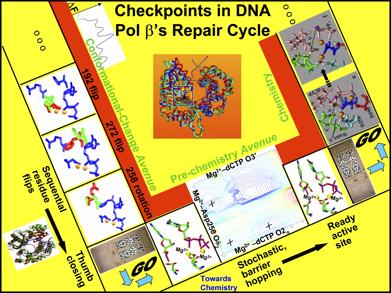
Figure 12. Sequential events and corresponding “gates” controlling pol β's fidelity (Radhakrishnan et al., 2006): the conformational change avenue, which comprises of Arg192 flip, Phe272 flip, and Arg258 rotation accompanying thumb subdomain closing motions upon incoming nucleotide binding, the pre-chemistry avenue, which involves the stochastic reorganization of the protein catalytic region, particularly the coordinating ligands of the two binding metal ions, and the chemistry avenue, where the incoming nucleotide is finally connected onto the primer terminus and the primer is extended by one residues.