Can Classical Equations Simulate Quantum-Mechanical Behavior? A Molecular
Dynamics Investigation of a Diatomic Molecule with Morse Potential
In a recent paper, we presented a new computational method for molecular
dynamics which uses the Backward-Euler scheme to solve the classical Langevin
dynamics equations. Parameters for the simulation include a target temperature
T, a time step  t, and a cutoff
frequency
t, and a cutoff
frequency 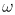 c. We
showed for a harmonic oscillator system that the cutoff frequency can be
set as c=kT/
c. We
showed for a harmonic oscillator system that the cutoff frequency can be
set as c=kT/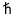 in order to mimic quantum-mechanical behavior. We now continue this investigation
for a nonlinear case: a diatomic molecule governed by a Morse bond potential.
Since approximate quantum-mechanical energy levels are explicitly known
for this model, a comparison of energies can be made with molecular dynamics
results. By performing dynamics runs for a wide range of temperatures and
calculating mean energies, we find a very good agreement between these
energies and quantum mechanical predictions. Vibrational excitation begins
at temperatures around 800 K, and for higher temperatures both energy curves
(molecular dynamics and quantum mechanics) approach the classical prediction
of
in order to mimic quantum-mechanical behavior. We now continue this investigation
for a nonlinear case: a diatomic molecule governed by a Morse bond potential.
Since approximate quantum-mechanical energy levels are explicitly known
for this model, a comparison of energies can be made with molecular dynamics
results. By performing dynamics runs for a wide range of temperatures and
calculating mean energies, we find a very good agreement between these
energies and quantum mechanical predictions. Vibrational excitation begins
at temperatures around 800 K, and for higher temperatures both energy curves
(molecular dynamics and quantum mechanics) approach the classical prediction
of 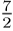 kT energy
per molecule. Future investigations will focus on more general nonlinear
potential functions employed in force fields of nucleic acids and proteins
kT energy
per molecule. Future investigations will focus on more general nonlinear
potential functions employed in force fields of nucleic acids and proteins

 Click to go back to the publication list
Click to go back to the publication list
Webpage design by Igor Zilberman