The 3 billion DNA base pairs in each nucleus of a human cell are densely packed within the chromatin fiber, a nucleoprotein complex in which the DNA wraps around nucleosomes much like yarn around many spools (Fig. 1). The tightly-packed and organized structure of the chromatin fiber in eukaryotes serves two essential cellular functions: it condenses meters-long genomic DNA by several orders of magnitude to enable its packaging into micrometer-sized cell nucleus, and it regulates the template-directed transcription of genes through local unfolding of chromatin. With recent discoveries linking nucleosome positioning to gene activity and higher order genome organization, the need to understand chromatin organization has intensified, because the structural puzzle that explains genome organization underlies the most basic cellular functions, including transcription activation, gene silencing, and epigenetic control.
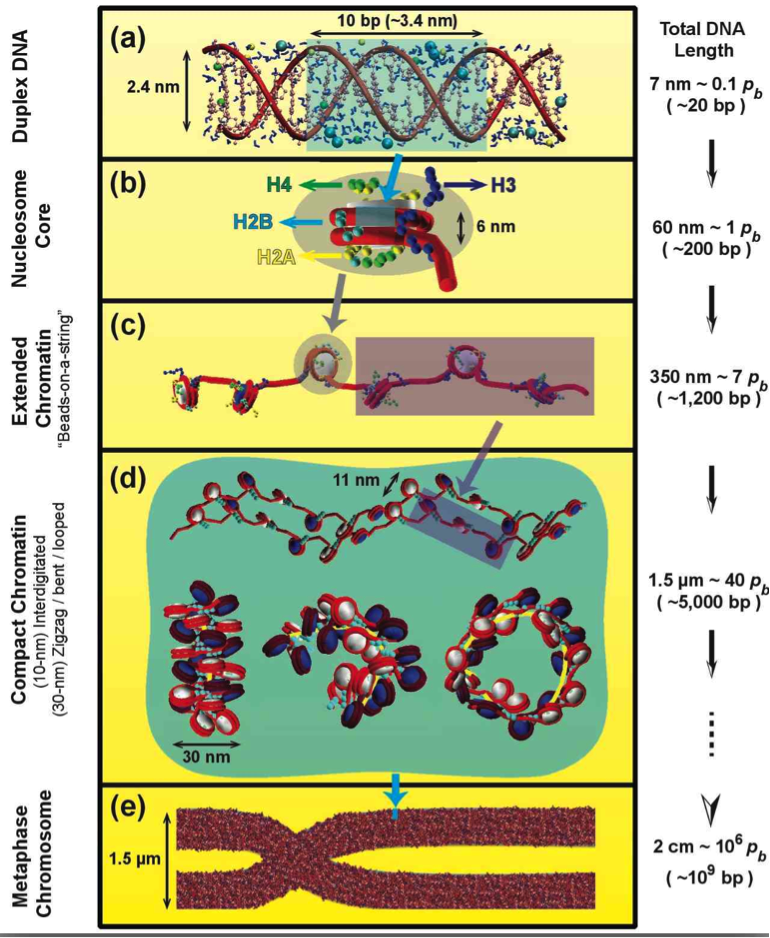
Fig. 1: Eukaryotic DNA in the cell.
In eukaryotes, the DNA is organized in the chromatin fiber, a complex made of DNA wrapped around core of 8 proteins, 2 copies each of H2A, H2B, H3, and H4. The chromatin building block is the nucleosome. At low salt, the fiber forms “beads-on-a-string” like models and, at physiological salt concentrations, it adopts more compact states, whose detailed structures are unknown. More condensed fiber structures form during transcription-silent phases of the cell cycle.
The solution to the “DNA folding problem” at different levels of folding under various cellular conditions can be considered more challenging than the protein folding problem because it occurs on a much greater range of spatial scales involving the entire genome (see Fig. 2).
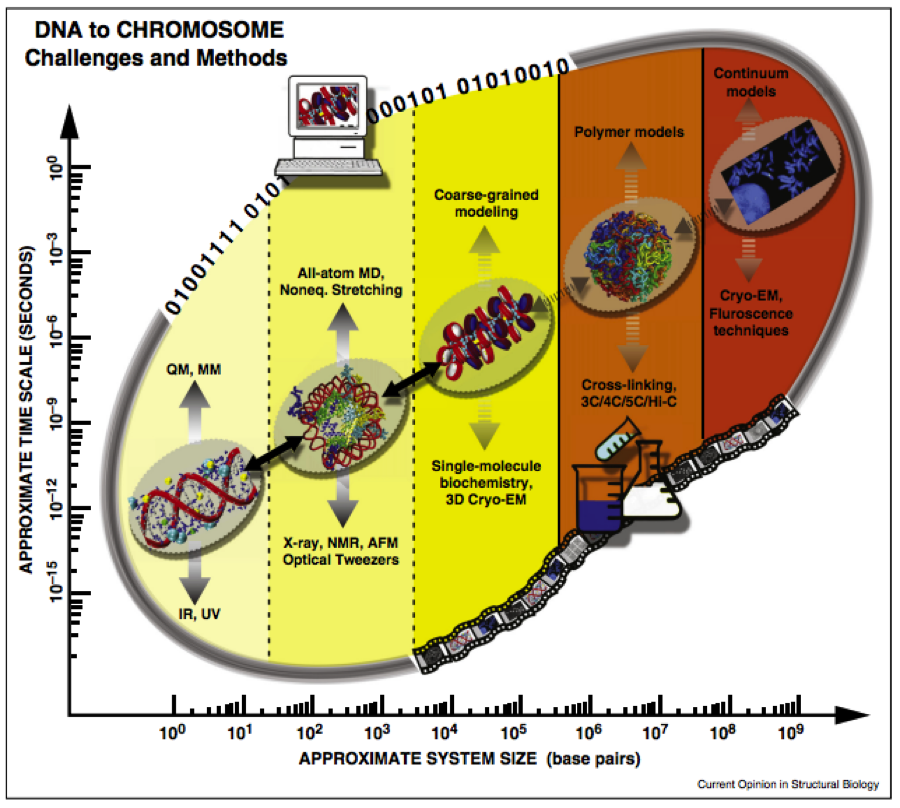
Fig. 2: The DNA folding problem is spans multiple time and space scales, which can be studied by both experiment and computational methods (Ozer, Luque & Schlick 2015).
The detailed structure of the chromatin fiber has remained controversial for over three decades, but the emerging view has been that fluid and heterogenous zigzag dominant fibers or polymer melt configurations with variable diameters are more relevant in the natural cellular environment.
Over several years, we have developed and applied chromatin models in increasing level of detail, each of which is suitable for certain biological problems and applications. Our first-generation “macroscopic” models treated the nucleosome and the wound DNA according to general mechanical and electrostatic properties, with the histone tails approximated as rigid bodies and linker histones neglected (Beard and Schlick, 2001a; Beard and Schlick, 2001b; Beard and Schlick, 2003; Zhang et al., 2003; Sun et al., 2005). It captured the fundamental monovalent-salt dependent mechanics of chromatin and the thermal fluctuations of the nucleosome and linker DNA.
Our next-generation mesoscale model (Sun et al., 2005; Arya et al., 2006; Arya and Schlick, 2006; Zhang et al., 2003) accounted for the irregular surface of the nucleosome, flexibility of the histone tails, presence of linker histone, and divalent ion effects (to a first-order approximation); see Fig. 3. Our current model has been carefully parameterized using available experimental data and thoroughly tested to reproduce energetic and dynamic data for oligonucleosomes, as summarized in (Arya and Schlick, 2009). We also incorporate variable linker lengths (Collepardo-Guevara & Schlick 2011), dynamic and static linker histone binding (Collepardo-Guevara & Schlick 2012), and histone tail acetylation (Collepardo-Guevara et al., 2015), various linker histone variants (Ognjen et al., 2019), other protein binding (Mao et al., 2023), and realistic values of nucleosome positions and other epigenetic regulators (Bascom et al., 2017; Gomez-Garcia et al., 2021; Yusufova et al., 2021; Swygert et al., 2021) for both Monte Carlo sampling and Brownian Dynamics (Li et al., 2023).
Our recent work focuses on gene folding at nucleosome resolution to integrate structural and dynamical aspects of chromatin organization to delineate the mechanisms of transcriptional regulation mediated through protein factors, epigenetic marks, and environmental conditions. Of particular interest are aspects of epigenetic regulation associated with human diseases such as cancers.
The nucleosome core is modeled as an irregular-shaped rigid body with pseudo charges on it surface. Linker DNA is modeled as beads using the discrete version of the worm-like chain model. The linker histones H1c and H1d are represented by 27/28 coarse grained beads with several binding modes (Perisic et al. 2019), and the histone tails are treated using the protein bead model where 1 bead represents 5 amino acids. For a full treatment, see (Bascom and Schlick 2017).
Recent Reviews
The chromatin fiber: multiscale problems and approaches
Genome modeling: From chromatin fibers to genes
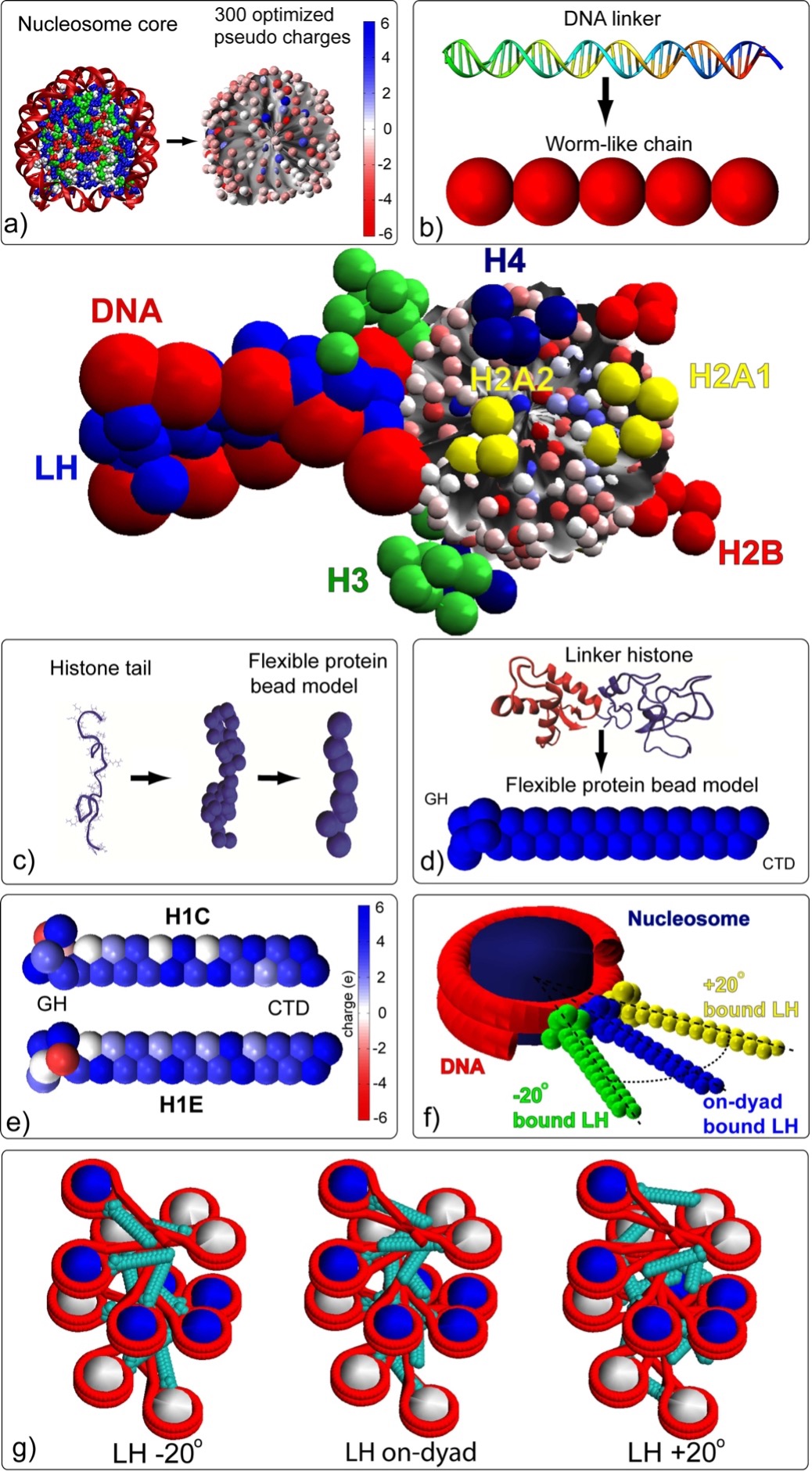
Fig. 3: Mesoscale oligonucleosome model.
The nucleosome core is modeled as an irregular-shaped rigid body with pseudo charges on it surface. Linker DNA is modeled as beads using the discrete version of the worm-like chain model. The linker histone is represented by 28 coarse grained beads, and the histone tails are treated using the protein bead model where 1 bead represents 5 amino acids. For a full treatment, see (Bascom and Schlick 2017).